
A urine pregnancy test device is an In-Vitro Diagnostic (IVD) hormonal assay that measures the beta (β) subunit of Human Chorionic Gonadotrophin (hCG) levels in urine. It is also known as a ‘home pregnancy test’, an ‘over-the-counter (OTC) pregnancy test’, or a ‘point-of-care pregnancy test’ if a provider administers the test. The generic device is designed to provide a binomial outcome or a positive or a negative result.
The medical device classification for pregnancy tests is based on the perceived risk associated with using the device. The class of the device within a regulatory authority will ultimately determine the choice of its testing pathways and the applicable quality standards and regulatory requirements. The U.S. FDA and Health Canada have classified pregnancy tests as a Class II device (medium risk), and applicable to “substantial equivalence” to marketed or predicate devices (510(k) clearance).
Quality Standards
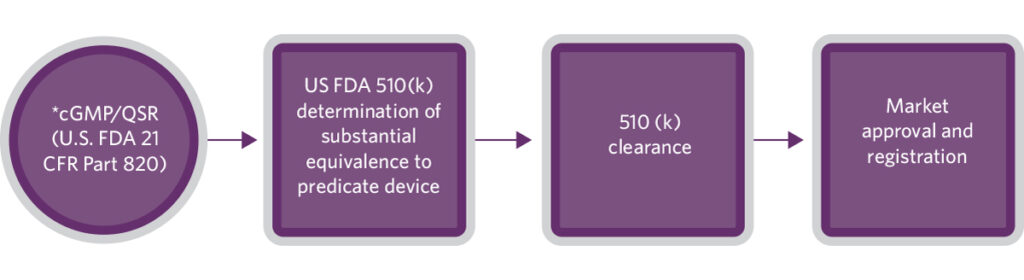
U.S. FDA governs the cGMP requirements for medical devices in part 820 (21 CFR Part 820) and incorporates them into a quality system regulation (QSR). The QSR includes requirements related to the methods, facilities, and controls used in the design, manufacturing, packaging, labeling, storing, and performance and maintenance of medical devices intended for human use. The global equivalent is ISO 13485.
For low-to medium-risk class products, compliance with quality-related regulations often depends on the manufacturer’s declaration. Manufacturers are expected to prepare QMS-controlled technical documentation including a description of product design and manufacturing process.
Note: The proof of industry standard for quality management, ISO 9001, is not satisfactory evidence for compliance with ISO 13485. All ISO 13485 requirements are specific to organizations providing medical devices and are required in many countries as the basis for quality assurance management of IVDs for their registration and regulatory control.
Regulatory Requirements
The level of regulatory scrutiny is determined by the level of the potential risks of medical devices. Since pregnancy tests are classified as class II IVD, a product that intends to launch in the United States market is required to submit a 510(k) application, also known as premarket notification.
‘Substantial equivalence’ is a part of the U.S. FDA 510(k) clearance process, where the manufacturer provides evidence indicating that the product to be marketed is at least as safe and effective as a marketed predicate device. FDA will evaluate the product’s intended use, performance, and labeling for clearance determinations. Upon the receipt of written notification of FDA clearance confirming ‘substantial equivalence’, the product can be marketed in the U.S.
See below for the required elements for FDA 510(k) application:
Below is a checklist of the requirements for premarket submissions for pregnancy tests intended for over-the-counter use:
Quality Standards
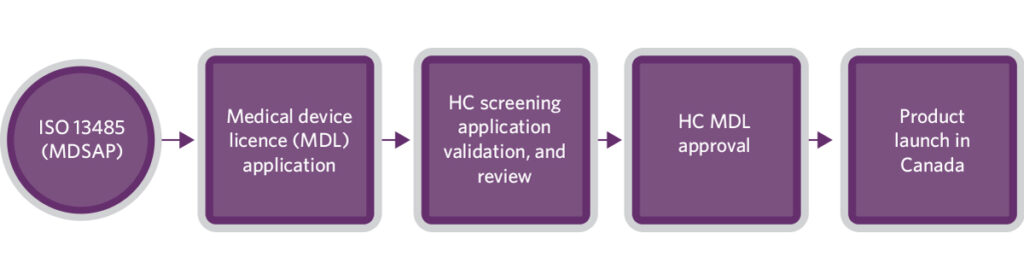
Health Canada (HC) requires medical device manufacturers to use a quality system certificate as evidence of compliance with the appropriate regulatory quality system requirement. HC only accepts the Medical Device Single Audit Program (MDSAP) certificate issued by Health Canada-authorized Conformity Assessment Bodies (CAB).
Regulatory Requirements
Since pregnancy tests are categorized as class II medical devices in Canada, a Medical Device Licence (MDL) approval by HC is required for a product to be marketed. For class II devices, the process of obtaining an MDL is usually faster than that for 510(k) clearance. There are different application forms for devices in each class. The submitted class II device application will undergo HC’s administrative screening, regulatory screening, administrative processing, and review process.
HC recommends all manufacturers use the guidance documents and tools available on their website when preparing and submitting the MDL application, as inapplicable, incomplete, or unacceptable applications can significantly delay or result in application rejection.
The pathway to market for your pregnancy test depends on the regulatory authority responsible for the market in which you launch your product. In the US, a pregnancy device is classified by the FDA as a class II medical device and requires a 510(k) application to go to market. In Canada, a pregnancy device is classified by Health Canada as a class II medical device and requires an MDL application in order to go to market.
Need assistance getting a pregnancy device to market in Canada or the United States? dicentra is a Contract Research Organization and Consulting firm which helps medical device companies bring their vital products to market. We have extensive regulatory and clinical experience with Medical Devices in Canada and the United States. We have provided clinical research support and consulting expertise to hundreds of medical device clients. Our combination of clinical and regulatory expertise specific to medical devices means we understand the full product lifecycle better than any of our competitors, to the benefit of our clients and sponsors.
Contact us for assistance in bringing your pregnancy test devices to market.