During the pandemic of respiratory disease caused by a novel coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) or Coronavirus Disease 2019 (COVID-19), the FDA Commissioner may authorize the use of unapproved medical products, or unapproved uses of approved medical products under Section 564 of the Federal Food, Drug, and Cosmetic Act (FD&C Act), given the current emergency circumstances imposed by COVID-19.
Amid a public health emergency, the FDA can issue an Emergency Authorization of Use (EUA) to provide accelerated assess to critical medical products when there are no adequate, approved, and available alternative options.
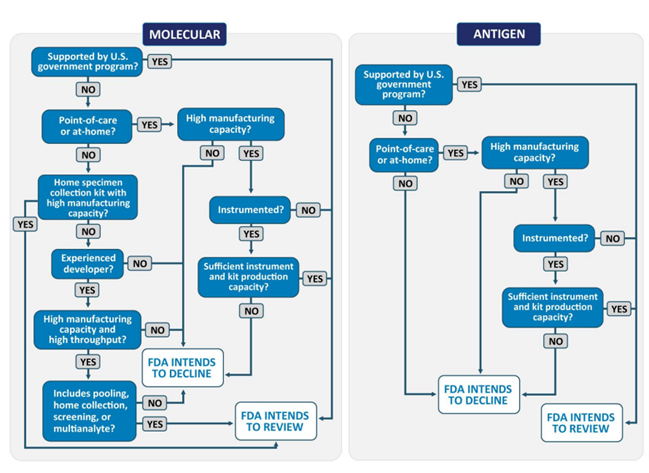
At this stage of the pandemic for SARS-CoV-2 diagnostic tests, the FDA focuses its efforts and prioritizes its reviews on tests that will significantly increase testing capacity and accessibility. For diagnostic tests (molecular and antigen), the FDA is focusing on tests that can be used at the Point-of-Care (POC) or completely at home from developers that have the capacity and the capability to scale up manufacturing shortly after authorization, preferably those with a manufacturing capacity of at least 500,000 tests per week, within the first 3 months of authorization. The same manufacturing capacity is required for developers requesting a EUA for laboratory-based molecular diagnostic tests. Laboratory-based molecular diagnostic tests that are highly sensitive with high throughput and are intended for pooling, home specimen collection, screening, or detection of multiple analysts, are also part of the scope of this prioritized review. The below flowcharts give a general overview of the FDA review of EUA requests for COVID-19.
The issuance of authorization under a EUA is evaluated on a case-by-case basis. In general, the FDA will assess the following criteria for compliance:
Applicants pursuing this pathway can utilize the available EUA templates as provided by the FDA to facilitate the preparation and submission of a EUA request for various types of COVID-19 tests. Note, devices authorized under EUA are only permitted to be marketed during the current public health emergency.
The De Novo premarket review pathway is a pathway for a new type of medical device that is of low to moderate risk and when there is no legally marketed predicate device. Applicants pursuing this regulatory pathway are subject to special controls for labeling and performance testing as well as general controls. When requirements for special controls and general controls are met, the safety and effectiveness of tests are considered to be assured.
On March 17, 2021, the FDA granted the first marketing authorization of a COVID diagnostic test for the simultaneous qualitative detection and identification of multiple respiratory viral and bacterial nucleic acids in nasopharyngeal swabs (NPS) using the De Novo Premarket Review Pathway. The approval of this diagnostic test creates a new regulatory pathway, that any devices that are of the same type and the same intended use may go through the FDA’s 510(k) pathway.
A 510(k) is a premarket submission to the FDA demonstrating that the device included is as safe and effective, and is substantially equivalent, to a legally marketed device as per Section 513 (i)(1)(A) of the FD&C Act. The applicant needs to compare the device with one or more legally marketed devices and substantiate the comparability.
On November 1, 2021, the FDA cleared the first 510(k) for a COVID-19 test, which grants this device authorization to market it beyond the public health emergency.
If you would like to get your COVID-19 test to market in the U.S contact us to speak to one of our medical device experts who can help get your device approved and to market.